

- 最新进展
- 产品信息
最新进展
USP7:抗肿瘤药物的新希望与变构小分子的突破
在癌症治疗的漫长征途中,科学家们一直在寻找能够精准打击肿瘤细胞的“武器”。近年来,一个名为USP7的靶点引起了广泛关注。USP7,又被称为HAUSP,是一种去泛素化酶(DUBs),它在细胞内扮演着调节蛋白稳定性的关键角色。然而,尽管USP7是一个极具潜力的抗肿瘤药物靶点,传统药物开发却一直面临着靶点选择性的瓶颈。直到最近,一项突破性的研究带来了新的曙光——首个针对USP7的变构小分子的发现。
USP7:抗肿瘤的关键靶点
USP7属于去泛素化酶家族,它的主要工作是去除蛋白质上的泛素链,从而调节蛋白质的稳定性和功能。在细胞中,USP7能够稳定多种与癌症发生密切相关的蛋白质,例如MDM2。MDM2是一种重要的负调控因子,它能够抑制p53蛋白的活性,而p53蛋白是细胞内重要的“肿瘤监视器”,能够启动细胞凋亡程序,阻止癌细胞的增殖。因此,通过抑制USP7的活性,可以促进MDM2的降解,恢复p53蛋白的功能,从而诱导肿瘤细胞凋亡,达到抗肿瘤的效果。
传统药物开发的困境
尽管USP7的抗肿瘤潜力早已被证实,但药物开发之路却并不平坦。去泛素化酶家族的成员在传统结合位点上具有高度保守性,这意味着针对这些位点开发的小分子药物往往缺乏足够的靶点选择性。换句话说,这些药物不仅会抑制USP7,还可能误伤其他家族成员,导致严重的副作用。过去十年中,尽管科学家们不断努力,开发出了一些更有效和更具选择性的USP7抑制剂,但始终未能突破这一瓶颈。
变构小分子的突破
就在人们几乎陷入僵局之时,Gavory等人的研究带来了新的希望。他们采用了一种全新的策略——基于结构的药物设计,通过组合第一代抑制剂和分子碎片库的方法,成功发现了首个针对USP7的变构小分子。这种小分子的作用位点位于USP7的变构结合位点,远离传统的催化位点。这一巧妙的设计使得小分子在细胞实验中能够选择性地与USP7相互作用,而不会与其他家族蛋白发生作用。实验结果令人振奋:在对38个人类去泛素化酶、63个其他蛋白酶和49个代表性激酶进行的体外试验中,该化合物对USP7靶点表现出极高的选择性。
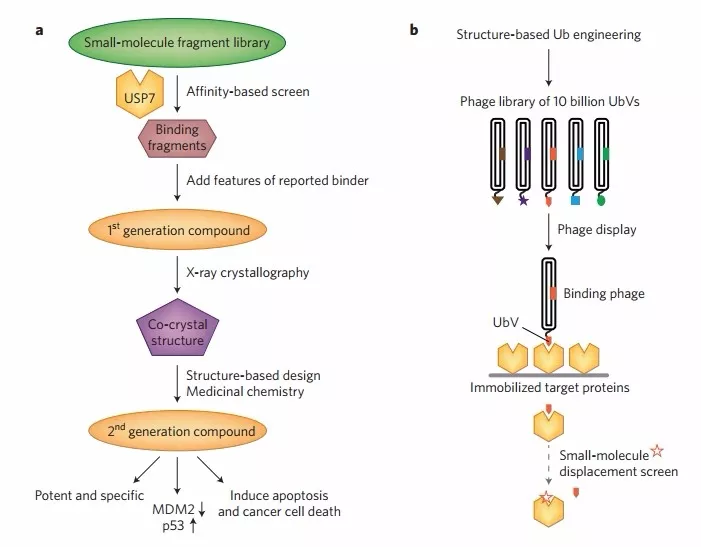
变构抑制机制的奥秘
这种变构小分子的成功,源于其独特的抑制机制。USP7蛋白在发挥功能时,其构象会发生变化,而这个变构小分子正是利用了这种变化的构象来发挥作用。虽然目前其具体的变构抑制机制还需要进一步的酶学实验和不同状态下的USP7蛋白晶体结构研究来阐明,但这一发现已经为USP7的药物开发开辟了新的道路。
未来展望
USP7变构抑制剂的发现不仅为抗肿瘤药物的研发带来了新的希望,也为其他具有高度保守性结合位点的蛋白家族的药物开发提供了新的思路。在去泛素化酶家族中,传统底物位点的保守性一直是药物开发的难题,而变构小分子的成功表明,通过探索蛋白的其他结合位点,或许可以找到更具选择性和活性的药物。未来,随着更多变构抑制剂的发现和研究的深入,我们有望开发出更加精准、高效的抗肿瘤药物,为癌症患者带来更多的福音。
USP7的故事还在继续,而变构小分子的突破只是一个开始。在科学的探索之路上,每一次小小的进步都可能带来巨大的变革。让我们期待更多创新的药物能够从实验室走向临床,为人类的健康保驾护航。
声明:本篇文章在创作中部分采用了人工智能辅助。如有任何内容涉及版权或知识产权问题,敬请告知,我们承诺将在第一时间核实并撤下。
买重组蛋白,找南京优爱
优爱蛋白专注于提供药物研发、细胞治疗、基因治疗、基础科研所需各种蛋白类试剂原材料和服务,包括药物靶点蛋白、免疫检查点蛋白、细胞因子、工具酶、 蛋白定制表达、全长跨膜蛋白开发等。优爱致力于为客户提供优质的产品和专业服务,打造具有国际竞争力的高新技术企业。
靶点蛋白 | 膜蛋白 | 细胞因子 | 酶 | 病毒抗原 | 蛋白定制
南京优爱生物科技研发有限公司 邮箱:order@ua-bio.com 热线:0571-87565022

产品信息

友情链接:






©2021-2026 南京优爱生物科技研发有限公司版权所有
网站备案号 浙ICP备2022019033号-2 浙公网安备33010202004867号
电话:0571-87565022
邮箱:order@ua-bio.com
本网站销售的所有产品均不得用于人类或动物之临床诊断或治疗,仅可用于工业或者科研等非医疗目的。
