- 最新进展
- 产品信息
酪氨酸激酶c-Met作为肝细胞生长因子(HGF)受体,在调控细胞增殖、迁移与血管生成中发挥核心作用。其信号通路的异常激活(包括基因扩增、突变或过表达)已被证实与肺癌、胃癌、肝癌等多种恶性肿瘤的进展和转移密切相关。尽管c-Met特异性抑制剂(如tepotinib、capmatinib)在临床治疗中展现出初始疗效,但获得性耐药(如D1228N、Y1230H等位点突变)的普遍出现严重限制了其长期应用。近年来,蛋白降解靶向嵌合体(PROTAC)技术通过诱导靶蛋白泛素化降解,为克服传统抑制剂耐药难题提供了新思路。然而,第一代c-Met靶向PROTAC分子仍面临口服生物利用度低、代谢稳定性差等关键技术瓶颈。

近期,国家应急防控药物工程技术研究中心、国家安全特需药品全国重点实验室联合中国医科大学研究团队在《Journal of Medicinal Chemistry》上发表了题为“Novel Highly Potent c-Met Degraders against a Broad Range of Cancers”的研究论文(DOI: 10.1021/acs.jmedchem.5c01470)。该研究通过系统性结构优化,成功开发出新一代c-Met降解剂D19、D26和G4,在保持高效降解活性的同时显著改善了药代动力学特性。其中先导化合物D19在多种临床前模型中表现出近乎完全的肿瘤抑制效果,且能有效克服耐药突变,为c-Met驱动型肿瘤的临床治疗提供了突破性解决方案。
一、如何通过理性设计突破PROTAC分子的成药性瓶颈?
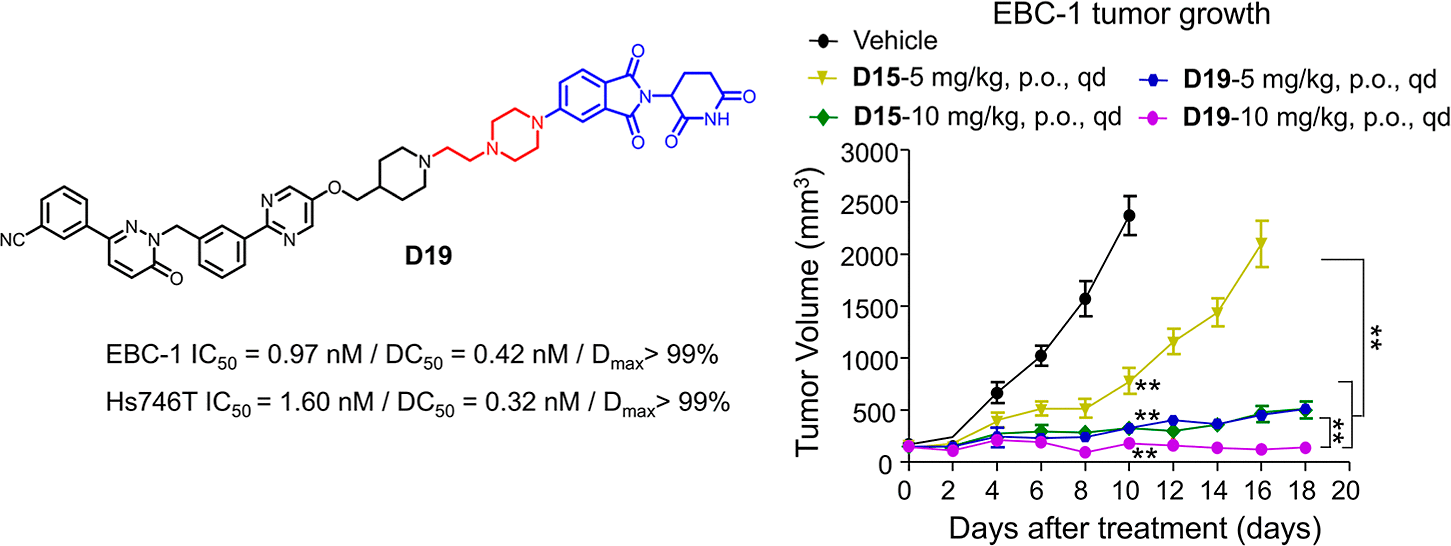
研究团队以前期开发的降解剂D15(Acta Pharm. Sin. B. 2023;13(6):2715-2735)为起点,针对其口服生物利用度低(0.7%)和代谢稳定性差的核心缺陷,实施了多维度的结构优化策略。首先,将连接链(linker)中的酰胺键替换为亚甲基单元,显著增强了对水解酶的稳定性,由此获得候选分子D19。其次,通过系统筛选E3连接酶配体,发现CRBN配体(沙利度胺衍生物)相比VHL配体更利于形成稳定的c-Met–PROTAC–CRBN三元复合物,据此优化得到降解剂D26。此外,团队还通过精简分子骨架开发出低分子量衍生物G4,在维持降解活性的同时显著改善类药性。这一系列基于结构生物学的精准修饰,为PROTAC技术的临床转化提供了重要的分子设计范式。
二、新型降解剂在体外模型中是否展现出超强效的抗肿瘤活性?
在细胞水平实验中,D19和G4对多种c-Met异常表达的癌细胞系(包括EBC-1非小细胞肺癌、Hs746T胃癌、MHCC97H肝癌等)表现出纳摩尔级的增殖抑制活性(IC₅₀ = 0.97–12.4 nM),显著优于前代分子D15及临床常用抑制剂tepotinib、capmatinib等。更值得注意的是,二者可通过CRL4CRBN E3连接酶介导的泛素-蛋白酶体途径,实现皮摩尔级的c-Met蛋白降解效率(DC₅₀ = 0.21–0.52 nM),最大降解率超过99%。Western blotting分析进一步证实,仅1 nM浓度的D19即可显著降低磷酸化c-Met(p-c-Met)及其下游信号分子STAT3的活化水平,10 nM时能完全阻断c-Met信号通路。尤为重要的是,降解剂对正常细胞(LO2肝细胞、293T肾上皮细胞)未表现出明显毒性(IC₅₀ > 100 μM),展现出优异的选择性。
三、优化后的降解剂能否解决药代动力学难题?
药代动力学评估显示,D19和G4在大鼠模型中的口服生物利用度分别达到7.54%和7.43%,较D15(0.3%)提升约20倍。同时,二者的半衰期(t₁/₂)延长至12.47小时和11.42小时,血浆暴露量(AUC0–∞)显著增加,且肝微粒体代谢稳定性大幅改善。这些数据表明,通过理性的连接链优化和分子精简策略,可有效突破PROTAC类分子常见的吸收障碍和快速代谢问题,为口服给药途径奠定了基础。
四、体内实验是否验证了其实际抗肿瘤效果与安全性?
在EBC-1异种移植瘤模型中,每日口服5 mg/kg剂量的D19即可抑制91.9%的肿瘤生长(TGI),10 mg/kg剂量组更达到98.4%的TGI,且部分实验动物实现肿瘤完全消退。值得注意的是,停药18天后未观察到肿瘤复发迹象。组织学分析显示,治疗组肿瘤细胞凋亡标志物(Cleaved Caspase-3)显著上调,增殖指标(Ki-67)明显抑制,而重要器官(心、肝、肾)未出现病理损伤,小鼠体重保持稳定,证明D19在高效抑瘤的同时具备良好的安全性。
五、新型降解剂能否克服临床耐药突变?
为验证其对耐药突变的抑制作用,研究团队构建了携带c-MetD1228N和c-MetY1230H点突变的细胞模型。结果显示,D19对D1228N和Y1230H突变株的IC₅₀分别为25.4 nM和40.8 nM,而临床抑制剂tepotinib在同等条件下的IC₅₀超过1000 nM。进一步机制研究表明,D19可在1000 nM浓度下完全降解突变型c-Met蛋白,并有效抑制其自磷酸化活性,这一特性显著优于仅能部分抑制激酶活性的传统ATP竞争性抑制剂。
六、该研究对PROTAC技术发展有何深远意义?
本研究不仅成功开发出具有临床转化潜力的c-Met降解剂,更在PROTAC分子设计策略上取得重要突破。首先,通过连接链刚性化和代谢位点屏蔽,解决了PROTAC口服吸收难题;其次,利用CRBN配体的协同效应增强降解效率;此外,低分子量衍生物G4的成功研制证明,在保持效力的同时优化类药性具有可行性。这些经验为针对其他激酶靶点的PROTAC开发提供了可借鉴的技术路径。
结论与展望
该研究通过多层次的理性设计,成功将c-Met靶向PROTAC分子的活性、稳定性和药代特性提升至新高度。先导化合物D19展现出的广谱抗肿瘤活性、克服耐药能力及口服有效性,标志着PROTAC技术向临床应用迈出关键一步。未来研究可进一步探索其在不同c-Met驱动肿瘤人群中的疗效差异,以及与其他免疫疗法或靶向药物的协同机制。随着更多临床前及临床数据的积累,这类降解剂有望为存在c-Met通路异常的患者提供更优治疗选择,并推动肿瘤靶向治疗进入“蛋白降解”新时代。
声明:本篇文章在创作中部分采用了人工智能辅助。如有任何内容涉及版权或知识产权问题,敬请告知,我们承诺将在第一时间核实并撤下。
买重组蛋白,找南京优爱
优爱蛋白专注于提供药物研发、细胞治疗、基因治疗、基础科研所需各种蛋白类试剂原材料和服务,包括药物靶点蛋白、免疫检查点蛋白、细胞因子、工具酶、 蛋白定制表达、全长跨膜蛋白开发等。优爱致力于为客户提供优质的产品和专业服务,打造具有国际竞争力的高新技术企业。